- Research
- Open access
- Published:
HMGB1/RAGE induces IL-17 expression to exaggerate inflammation in peripheral blood cells of hepatitis B patients
Journal of Translational Medicine volume 13, Article number: 310 (2015)
Abstract
Background
Hepatitis B (HB) is an infectious disease with unfavorable consequence for patients and involved in chronic inflammation of liver. The present study aimed to investigate whether High-mobility group protein B (HMGB)1/receptor for advanced glycation end products (RAGE) aggravates inflammation enhancing the expression of interleukin (IL)-17.
Methods
Mild and severe HB liver tissue and peripheral blood samples were obtained intra-operatively. Histological analysis of the livers was performed by immunohistochemistry. IL-1β and IL-6 of liver tissue were detected by confocal microscopy staining. Relative mRNA expression was measured by real-time PCR and protein levels were measured by enzyme-linked immunosorbent assay.
Results
HMGB1, RAGE and IL-17 expression is increased in liver of HB patients with acute on chronic liver failure (ACLF) compared to healthy controls. HMGB1 treatment induced inflammatory cytokines including IL-17 in peripheral blood cells of HB patients. IL-17 also induced the expression of RAGE and IL-1β in peripheral blood cells of HB patients with ACLF. On the other hands, the inhibitory factor of p38 and nuclear factor-kappa B reduced the expression of RAGE and IL-1β in peripheral blood cells HB patients with ACLF.
Conclusions
HMGB1, RAGE and IL-17 expression is increased in liver of severe HB patients. HMGB1 and RAGE interaction may contribute to the inflammation of liver enhancing the expression of IL-17, which can be possibly restored through the decline of the HMGB1/RAGE axis.
Background
Patients infected hepatitis B virus (HBV) which is a major human pathogen can be chronic hepatitis B or asymptomatic carrier, and reveal chronic inflammation of the liver associated with HBV. But, abnormal liver functions or liver failure can be initiated during protracted term of chronic HBV infection [1]. Some of HBV related with acute on chronic liver failure (ACLF) can induce rapid outbreak of inflammation, sickness with vomiting, cirrhosis and high mortality [2, 3]. Additionally, several proinflammatory cytokines are involved in liver inflammation because HBV leads to inflammatory response in liver. For example, tumor necrosis factor (TNF)-α is related with chronic HBV infection [4]. Additionally, it has been reported that the levels of TNF-α, IL-1β and IL-6 are induced by HBV infection in a human hepatocyte model [5].
High-mobility group protein B (HMGB) 1 is a cytokine mediator of inflammation and secreted by immune cells such as macrophages and monocytes. It is well documented that HMGB1 was secreted in activated macrophages and monocytes inducing inflammatory response [6]. HMGB1 is involved in liver inflammation enhancing proinflammatory cytokine such as IL-17 expression, which induces neutrophil infiltration and inflammatory response of liver [7]. It has been suggested that the severity of chronic HB is correlated with upregulated HMGB1 levels in [8]. In addition, HMGB1 increases the synthesis of pro-IL-1β in macrophages by the activation of p38 mitogen-activated protein kinase (MAPK) and nuclear factor kappa B (NF-κB) [9].
The receptor for advanced glycation end products (RAGE), which is transmembrane protein of the immunoglobulin super family, functions as a receptor for molecular pattern recognition activating innate and adaptive immunity. This receptor interacts with HMGB1 to promote inflammation. It has been reported that HMGB1/RAGE axis enhanced inflammation [10]. Moreover, it has been demonstrated that RAGE mediated the activation of p38 MAPK and NF-κB and induced the secretion of proinflammatory cytokines [11, 12].
IL-17 is a proinflammatory cytokines expressed by T-helper cells and conducts a significant role in inflammation. For example, IL-17 results in inflammatory response through activation of p38 MAPK and NF-κB [13]. There are several evidences that the expression of IL-17 is involved in several inflammatory diseases [14–16]. As IL-17 initiates immune response through upregulating chemokine production in several tissues such as liver, the expression of IL-17 is involved in HB. For example, interleukin (IL)-17 producing CD4+ T cells are involved in liver inflammation and also increased in the bloods of HB patients pervading the liver tissues of HB patients [17, 18]. It is also well documented that the expression of IL-17 was promoted in serum of patients with chronic HB and correlated with the severity of fibrosis [19].
We hypothesized that IL-17, HMGB1 and RAGE are increased in liver of HB. The current study attempts to determine whether HMGB1/RAGE axis induces the expression of IL-17 through the activation of p38 MAPK and NF-κb in peripheral blood cells patients with HB. Thus, this research was carried out to elucidate that the upregulation of IL-17 expression induced by HMGB1 and RAGE exaggerates inflammation in peripheral blood cells of HBV-related acute on chronic liver failure (ACLF) patients.
Methods
-
1.
Patients and clinical information.
We prospectively recruited eight patients with HBV-related ACLF at Seoul St. Mary’s Hospital (Seoul, Korea). Patients with comorbidities such as hepatitis C infection, human immunodeficiency virus infection, autoimmune liver diseases, hepatocellular carcinoma, alcoholic liver disease, and concurrent bacterial infection were excluded. Eight patients with HBV-related ACLF and eight healthy donors were age-matched and sex-matched, and then enrolled as controls for blood samples. The study was approved by the Institutional Review Board of Seoul St Mary’s Hospital (KC14SISI0008), and written informed consent was obtained from each subject. The patients were divided the severity of chronic hepatitis according to the Knodell Histology Activity Index Score [20]. Eight acute on chronic liver failure patients were enrolled in this investigation. Demographics of each acute on chronic liver failure patient group are listed in Table 1.
-
2.
Collection of peripheral blood mononuclear cells.
Peripheral blood mononuclear cells were isolated from heparinized blood samples by Ficoll-Hypaque (GE Healthcare, Bioscience, Sweden, Uppsala) density-gradient centrifugation, The isolated cells were cultured. Cell cultures were performed in RPMI1640 medium (GibcoBRL, Carlsbad, CA, USA) containing penicillin (100 U/Ml), streptomycin (100 μ/ml) and 10 % fetal bovine serum (GibcoBRL, Carlsbad, CA, USA) that had been inactivated by heating to 55 °C for 30 min. The cell suspensions were dispensed into 48-well plates (Nunc, Roskilde, Denmark).
-
3.
Cell treatment.
Peripheral blood mononuclear cells isolated from heparinized blood samples were treated by NF-κB inhibitor (Sigma), IL-17 (R&D Systems), p38 and sRAGE (InvivoGen).
-
4.
Measurement of cytokines.
Cytokines concentration of IL-1β, IL-6, TNF-α and IL-17 in culture supernatants were analysis by ELISA. Antibodies directed against human IL-1β, IL-6, TNF-α and IL-17 and against biotinylated anti-human IL-1β, IL-6, TNF-α and IL-17 were used as the capture and detection antibodies, respectively.
Alkaline phosphatase (Sigma) was used for the chromogenic reaction. The amounts of cytokines present in the test samples were determined from standard curves constructed with serial dilutions of recombinant human IL-1β, IL-6, TNF-α and IL-17 (R&D Systems, Minneapolis, MN, USA). The absorbance was determined with an ELISA microplate reader at 405 nm.
-
5.
RNA extraction and real-time quantitative polymerase chain reaction.
After incubation for 24 or 72 h with HMGB (R&D Systems, Minneapolis, MN, USA), mRNA was extracted using RNAzolB (Biotex Laboratories, Huston, TX, USA) according to the manufacturer’s instructions. Polymerase chain reaction (PCR) amplification and analysis were achieved using a LightCycler 2.0 instrument (Roche Diagnostic, Mannheim, Germany) with software version 4.0. All reactions were performed with the LightCyclerFastStart DNA Master SYBR green I (Takara, Shiga, Japan) according to the manufacturer’s instruction. The following primers were used: Human IL-17, 5′-CAA CCG ATC CAC CTC ACC TT-3′ (sense) and 5′-GGC ACT TTG CCT CCC AGA T-3′ (antisense); IL-6, 5′-TGC TCC TGG TGT TGC CTG CT-3′ (sense) and 5′-AGC CAC TGG TTC TGT GCC TGC-3′ (antisense); IL-1b, 5′-GGA CAA GCT GAG GAA GAT GC-3′ (sense) and 5′-TCG TTA TCC CAT GTG TCG AA-3′ (antisense); TNF-a, 5′-GCC TCT TCT CCT TCC TGA TCG T-3′ (sense) and 5′-CTC GGC AAA GTC GAG ATA GTC G-3′ (antisense); RAGE, 5′-GAC TCT TAG CTG GCA CTT GGA T-3′(sense) and 5′-GGA CTT CAC AGG TCA GGG TTA C-3′ (antisense). mRNA expression was normalized to β-actin expression.
-
6.
Immunohistochemical staining.
Liver tissue were fixed in 4 % paraformaldehyde and embedded in Paraffin. Then 7 μm sections were prepared. The sections were deparaffinized using xylene and dehydrated in a gradient of alcohols. Endogenous peroxidase activity was quenched with methanol 3 % H2O2. Immunohistochemistry was performed using the Vectastain ABC kit (Vector Laboratories, Burlingame, CA, USA). Tissue were incubated with the first primary antibody for IL-17 (Santacruz), HMGB (Cell signaling), RAGE (R and D systems), and isotype control overnight at 4 °C, and a biotinylated secondary linking antibody and a streptavidin peroxidase complex for 1 h. The final color product was developed using DAB chromogen (DAKO, Carpinteria, CA, USA).
-
7.
Immunofluorescence analysis.
A 100 μl aliquot of each sample was put into the appropriate well of a cytospin chamber (Thermo Scientific, Michigan, MI, USA) and was centrifuged art 800g for 3 min at 4 °C. To preserve the membrane-associated components and foreclose cytoplasmic staining, cells were fixed with methanol-acetone at −20 °C for 10 min. Immunofluorescence investigation were carried out using the PE-conjugated anti-IL-b and IL-6 (1:100; BD Bioscience) and 4′,6-diamidino-2-phenylindole (DAPI). The stained sections were analyzed using a Zeiss microscope (LSM 510 Meta; Carl Zeiss, Oberkochen, Germany) at 400× magnification.
-
8.
Statistical analysis.
Results were expressed as medians (interquartile range), Statistical comparison between two groups were made by a Mann–Whitney U test. A P < 0.05 was considered statistical significant. All statistical tests were performed using GraphPad Prism v.4.03 software (GraphPad Software, La Jolla, CA, USA).
Results
-
1.
Histological expression of HMGB1, RAGE and IL-17 in the liver of patients of different groups.
The expression of HMGB1, RAGE and IL-17 in liver tissues of HB patients was evaluated by histological analysis. These expressions were increased in liver tissues of severe HB patients compared to mild HB patients and healthy controls (Fig. 1a). Gene expression of HMGB1, RAGE and IL-17 was promoted significantly in peripheral blood mononuclear cells (PBMC) of HB patients (Fig. 1b).
Fig. 1 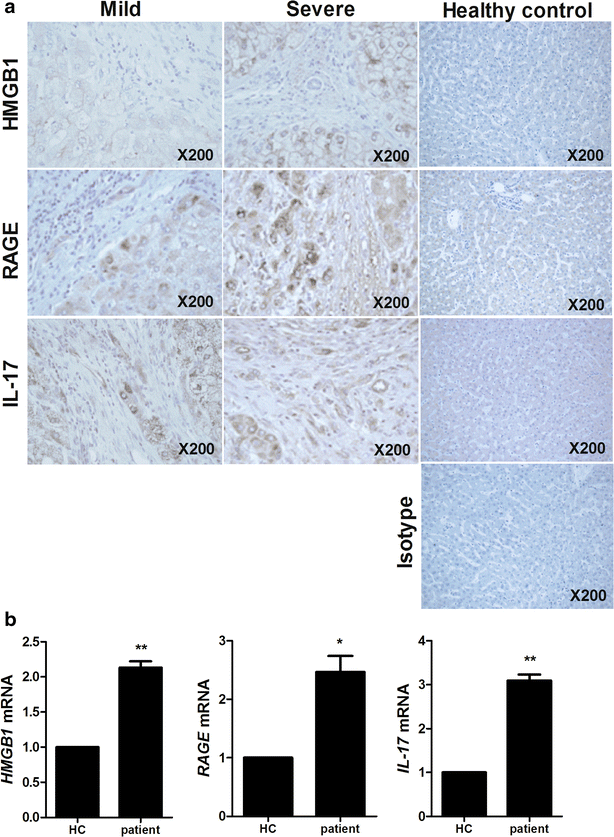
The expression of HMGB1, RAGE and IL-17 is positively related with severity of HB. a Histopathological analysis showed the expression of HMGB1, RAGE and IL-17 in liver of healthy controls, mild HB patients and severe HB patients. All tissues were counterstained with hematoxylin (original magnification, ×200). All images were obtained for each patients showing representative images. b The mRNA level of HMGB1, RAGE and IL-17 in peripheral blood cells of patients with HB was measured by realtime-PCR. Data are presented as the mean ± SD of three independent experiments (*p < 0.05, **p < 0.01)

-
2.
The expression of proinflammatory cytokines induced by HMGB1 treatment in PBMC of healthy controls and HB patients.
In order to determine whether HMGB1 can induce the inflammatory response in PBMCof healthy controls and HB patients, cells were cultured in the presence HMGB1. Real-time PCR was performed to investigate the effects of HMGB treatment (500 ng/ml) on the mRNA expression of proinflammatory (IL-1β, -6, -17 and TNF-α). The expression of proinflammatory cytokines in PBMC of HB patients were promoted significantly compared to healthy controls. Particularly, IL-17 production was increased time-dependently (Fig. 2a). Moreover, ELISA was used to measure the protein level of proinflammatory cytokines (IL-1β, -6, -17 and TNF-α) and these proinflammatory cytokines productions were increased significantly by HMGB1 treatment (500 ng/ml) compared to healthy controls (Fig. 2b).
Fig. 2 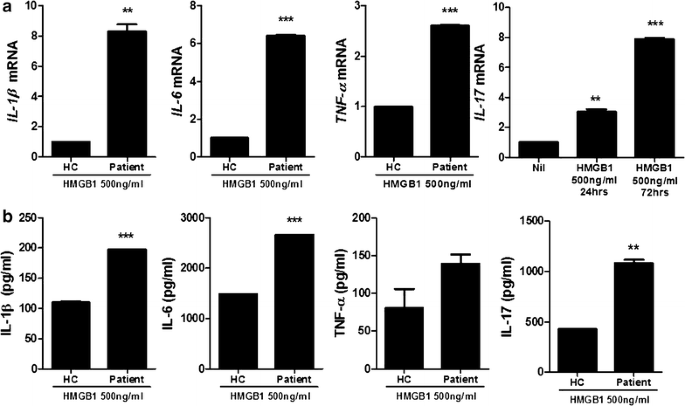
HMGB1 treatment promotes significantly the expression of proinflammatory cytokine in peripheral blood cells of HB patients compared to healthy controls. a The mRNA expression of IL-1β, -6, -17 and TNF-α in peripheral blood cells of patients with HB and healthy controls was measured by realtime-PCR (n = 3). b The protein level of IL-1β, -6, -17 and TNF-α in peripheral blood cells of patients with HB and healthy controls was measured by ELISA. Data are presented as the mean ± SD of three independent experiments (n = 3, **P < 0.03, ***P < 0.001)

-
3.
Immunocytochemical expression of proinflammatory cytokines in PBMC of healthy controls and patients with HB.
Confocal scanning revealed that IL-1β and -6 expressions were increased by HMGB1 treatment (500 ng/ml) in PBMC of HB patients (Fig. 3a, b). However, the expression of IL-1β and -6 in PBMC of healthy controls was not increased by HMGB1 treatment.
Fig. 3 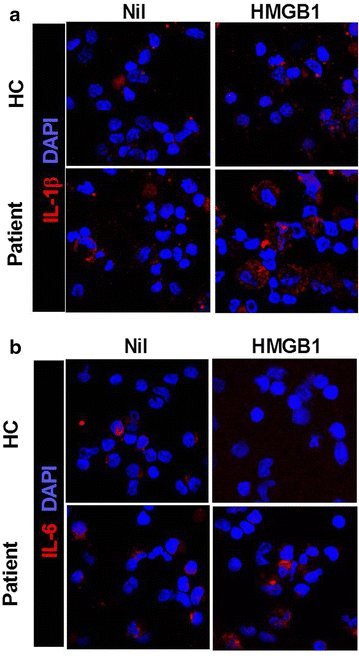
HMGB1 treatment more enhances the expression of proinflammatory cytokine in peripheral blood cells of HB patients than peripheral blood cells of healthy controls. Liver of HB patients or healthy controls as control were subjected to immunostaining for IL-1β and -6 (n = 3) (a, b)

-
4.
The expression of IL-17 induced by HMGB1 through RAGE.
Real-time PCR and ELISA were performed to investigate the variation of IL-17 expression in PBMC of HB patients. The PBMC was cultured with HMGB1 to examine that HMGB1 can induce IL-17 expression. HMGB1 treatment (500 ng/ml, 72 h) increased significantly mRNA expression of IL-17 in PBMC of HB patients. However, IL-17 gene expression is not enhanced in PBMC cultured with HMGB1 and soluble form of the receptor for advanced glycation end products (sRAGE) (Fig. 4a). In ELISA, IL-17 production is upregulated by HMGB1 treatment (500 ng/ml, 72 h) (Fig. 4b). But, the expression of IL-17 is not increased by the treatment of HMGB1 and sRAGE.
Fig. 4 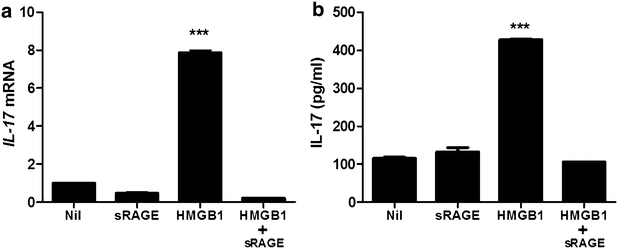
HMGB1 increases IL-17 expression through RAGE. The mRNA expression of IL-17 in peripheral blood cells of patients with HB was evaluated by realtime-PCR (a). The protein level of IL-17 in peripheral blood cells of patients with HB was evaluated by ELISA (b). Data are presented as the mean ± SD of three independent experiments (***p < 0.001)

-
5.
Inflammatory response induced.
To determine the induction of IL-1β and RAGE by IL-17, PBMC of HB patients was treated with IL-17 (10 ng/ml, 72 h). IL-17 treatment increased significantly the mRNA expression of RAGE and IL-1β in PBMC of HB patients. The expression of IL-1β in PBMC of HB patients was also promoted significantly by IL-17 (Fig. 5a, b). The inhibitory of p38 MAPK (10 μM, 72 h) and NF-κB (50 μM, 72 h) decreased markedly RAGE and IL-1β mRNA levels β in PBMC of HB patients (Fig. 5c).
Fig. 5 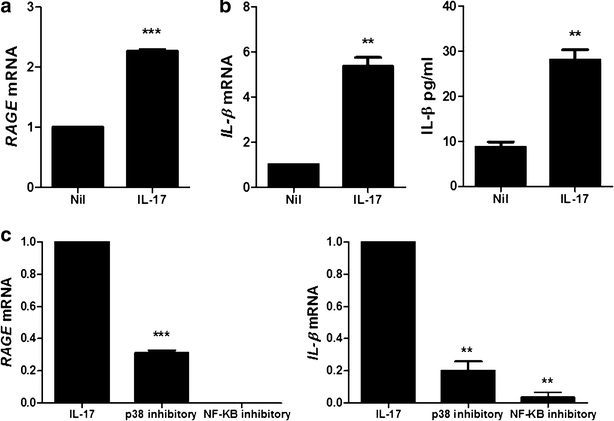
IL-17 promotes the expression of IL-1β and RAGE through p38 MAPK and NF-κB. a The mRNA expression of RAGE in peripheral blood cells of patients with HB was measured by realtime-PCR. b The mRNA expression and protein level of IL-1β in peripheral blood cells of patients with HB is measured by realtime-PCR and ELISA. c The mRNA expression of RAGE and IL-1β in peripheral blood cells of patients with HB was measured by realtime-PCR. Data are presented as the mean ± SD of three independent experiments (**P < 0.03, ***p < 0.001)

Discussion
It is generally believed that HMGB1, a significant chromatin protein interacting transcription factors, nucleosomes and histones, interacts with RAGE in inflammatory response and IL-17 cause inflammation through activation of p38 MAPK and NF-κB [13, 21–23]. However, there is a little evidence of the interaction of HMGB1/RAGE and IL-17 on inflammatory response and the mechanism of its action. Here, we studied the inflammatory activity of HMBG1/RAGE on inflamed peripheral blood cells through previously undiscovered mechanism.
The most meaningful observation in this study is reciprocal activity of HMGB1/RAGE and IL-17 in peripheral blood cells of HB patients. It has been demonstrated that the HMGB1/RAGE interaction may be pivotal in liver inflammation [24]. Several reports have indicated that IL-17 plays an important role in liver inflammation [25, 26]. But, the interaction of HMGB1/RAGE with IL-17 has been not investigated in liver inflammation with HB. In this investigation, we revealed the inflammatory function of HMGB1/RAGE inducing IL-17 production in peripheral blood cells of patients with HB.
Previous evidences have documented that HMGB1, RAGE and IL-17 are involved in liver inflammation [27–29]. We observed that the expression of HMGB1, RAGE and IL-17 in liver of severe HB patients is higher than these expressions in liver of mild HB patients. These results suggest that the expression of HMGB1, RAGE and IL-17 is positively related with severity of HB.
Several proinflammatory cytokines are involved in HB pathogenesis. It is well reported that IL-1β, -6 and TNF-α were increased in serum of HB patients [30]. In this study, HMGB1 treatment enhanced the gene expressions and protein levels of these cytokines in peripheral blood cells of HB patients compared to healthy controls. Thus, HMGB1 can aggravate inflammatory response in PB patients.
Even though HMGB1 and IL-17, inducer of inflammation, are involved in liver inflammation, there is a little evidence of the expression of IL-17 induced by HMGB1. Indeed, IL-17 expressing cells such as helper T cells and neutrophils are located in liver and involved in inflammatory liver disease [31, 32]. HMGB1 and RAGE are also expressed in human liver cells including Hepg2 cells and related with liver disorders such as hepatic injury and liver ischemia [24, 27, 33, 34]. In this investigation, IL-17 expression was promoted by HMGB1 treatment in peripheral blood cells of patients with HB. We also observed that HMGB1 leads to increase the expression of IL-17 through RAGE.
NF-κB, a key regulator in the immune response, and p38 MAPK are involved in inflammation. The activation of NF-κB and p38 MAPK enhanced the expression of inflammatory cytokine and is related with several inflammatory diseases including HB [35–37]. The noticeable finding is that IL-17 induces the mRNA level of RAGE and IL-1β expression and the inhibitor of p38 MAPK and NF-κB suppressed the mRNA expression of RAGE and IL-1β in peripheral blood cells of patients with HB. On basis of these results, we presumed that IL-17 may cause the expression of RAGE and IL-1β by the activation of p38 MAPK and NF-κB.
Although HMGB1 is potential to induce IL-17 expression and exaggerates HB, in vivo animal investigations are needed to confirm the inflammatory effect of HMGB1 treatment. In vivo animal studies conducted in HB model are required to further proof that HMGB1 results in the exaggeration of HB enhancing IL-17 expression. Moreover, in vitro assays covering upregulation of proinflammatory cytokines through HMGB1 treatment were performed using relatively small number of samples and, thus, showed the pilot data. However, this investigation is the first research to report and propose the possible pathogenic potential of attenuation of HMGB1 activity in HB patients with ACLF. Future studies with the large number of cases and in vivo animal experiments are thought to be required to verify our hypothesis more precisely.
The function of HMGB1 has been little studied in inflammatory response mediated by IL-17. The suppression of HMGB1 production established in this investigation indicates that HMGB1 promotes IL-17 expression and inflammation in HB. Our results demonstrate that the inhibition of HMGB1/RAGE interaction can reduce inflammation in HB. This prior investigation about HMGB1 inducing IL-17 suggests that HMGB1 can be strong therapeutic target in HB.
Conclusions
The function of HMGB1 has been little studied in inflammatory response mediated by IL-17. The suppression of HMGB1 production established in this investigation indicates that HMGB1 can promote IL-17 expression and inflammation in HB. Our results demonstrate that the inhibition of HMGB1/RAGE interaction can reduce inflammation in HB. This prior investigation about HMGB1 inducing IL-17 suggests that HMGB1 can be strong therapeutic target in HB.
References
Merle P, Trepo C, Zoulim F. Current management strategies for hepatitis B in the elderly. Drugs Aging. 2001;18(10):725–35.
Ganem D, Prince AM. Hepatitis B virus infection-natural history and clinical consequences. N Engl J Med. 2004;350(11):1118–29.
Jalan R, et al. Acute-on chronic liver failure. J Hepatol. 2012;57(6):1336–48.
Hohler T, et al. A tumor necrosis factor-alpha (TNF-alpha) promoter polymorphism is associated with chronic hepatitis B infection. Clin Exp Immunol. 1998;111(3):579–82.
Hosel M, et al. Not interferon, but interleukin-6 controls early gene expression in hepatitis B virus infection. Hepatology. 2009;50(6):1773–82.
Wang H, et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285(5425):248–51.
Wang X, et al. High-mobility group box 1 (HMGB1)-Toll-like receptor (TLR)4-interleukin (IL)-23-IL-17A axis in drug-induced damage-associated lethal hepatitis: interaction of gammadelta T cells with macrophages. Hepatology. 2013;57(1):373–84.
Liu HB, et al. Serum level of HMGB1 in patients with hepatitis B and its clinical significance. Zhonghua Gan Zang Bing Za Zhi. 2007;15(11):812–5.
He Q, et al. HMGB1 promotes the synthesis of pro-IL-1beta and pro-IL-18 by activation of p38 MAPK and NF-kappaB through receptors for advanced glycation end-products in macrophages. Asian Pac J Cancer Prev. 2012;13(4):1365–70.
Kang R, et al. The HMGB1/RAGE inflammatory pathway promotes pancreatic tumor growth by regulating mitochondrial bioenergetics. Oncogene. 2014;33(5):567–77.
Yeh CH, et al. Requirement for p38 and p44/p42 mitogen-activated protein kinases in RAGE-mediated nuclear factor-kappaB transcriptional activation and cytokine secretion. Diabetes. 2001;50(6):1495–504.
Zhu P, et al. Involvement of RAGE, MAPK and NF-kappaB pathways in AGEs-induced MMP-9 activation in HaCaT keratinocytes. Exp Dermatol. 2012;21(2):123–9.
Xie S, et al. IL-17 activates the canonical NF-kappaB signaling pathway in autoimmune B cells of BXD2 mice to upregulate the expression of regulators of G-protein signaling 16. J Immunol. 2010;184(5):2289–96.
Hashmi S, Zeng QT. Role of interleukin-17 and interleukin-17-induced cytokines interleukin-6 and interleukin-8 in unstable coronary artery disease. Coron Artery Dis. 2006;17(8):699–706.
Chen Z, O’Shea JJ. Th17 cells: a new fate for differentiating helper T cells. Immunol Res. 2008;41(2):87–102.
Song C, et al. IL-17-producing alveolar macrophages mediate allergic lung inflammation related to asthma. J Immunol. 2008;181(9):6117–24.
Zhang JY, et al. Interleukin-17-producing CD4(+) T cells increase with severity of liver damage in patients with chronic hepatitis B. Hepatology. 2010;51(1):81–91.
Zhang GL, et al. High level of IL-27 positively correlated with Th17 cells may indicate liver injury in patients infected with HBV. Liver Int. 2014;34(2):266–73.
Du WJ, et al. Expression of interleukin-17 associated with disease progression and liver fibrosis with hepatitis B virus infection: IL-17 in HBV infection. Diagn Pathol. 2013;8:40.
Knodell RG, et al. Formulation and application of a numerical scoring system for assessing histological activity in asymptomatic chronic active hepatitis. Hepatology. 1981;1(5):431–5.
Bianchi ME, Agresti A. HMG proteins: dynamic players in gene regulation and differentiation. Curr Opin Genet Dev. 2005;15(5):496–506.
Klune JR, et al. HMGB1: endogenous danger signaling. Mol Med. 2008;14(7–8):476–84.
He ZW, et al. HMGB1 acts in synergy with lipopolysaccharide in activating rheumatoid synovial fibroblasts via p38 MAPK and NF-kappaB signaling pathways. Mediat Inflamm. 2013;2013:596–716.
Basta G, et al. What is the role of the receptor for advanced glycation end products-ligand axis in liver injury? Liver Transpl. 2011;17(6):633–40.
Nagata T, et al. Requirement of IL-17RA in Con A induced hepatitis and negative regulation of IL-17 production in mouse T cells. J Immunol. 2008;181(11):7473–9.
Lafdil F, et al. Myeloid STAT3 inhibits T cell-mediated hepatitis by regulating T helper 1 cytokine and interleukin-17 production. Gastroenterology. 2009;137(6): 2125–35 e1–2.
Tsung A, et al. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J Exp Med. 2005;201(7):1135–43.
Pusterla T, et al. Receptor for advanced glycation endproducts (RAGE) is a key regulator of oval cell activation and inflammation-associated liver carcinogenesis in mice. Hepatology. 2013;58(1):363–73.
Meng F, et al. Interleukin-17 signaling in inflammatory, Kupffer cells, and hepatic stellate cells exacerbates liver fibrosis in mice. Gastroenterology. 2012;143(3):765–76 e1–3.
Torre D, et al. Serum levels of interleukin-1 alpha, interleukin-1 beta, interleukin-6, and tumor necrosis factor in patients with acute viral hepatitis. Clin Infect Dis. 1994;18(2):194–8.
Lemmers A, et al. The interleukin-17 pathway is involved in human alcoholic liver disease. Hepatology. 2009;49(2):646–57.
Wang L, Chen S, Xu K. IL-17 expression is correlated with hepatitis Brelated liver diseases and fibrosis. Int J Mol Med. 2011;27(3):385–92.
Huang ZB, et al. HMGB1 release by human liver L02 and HepG2 cells induced by lipopolysaccharide. Mol Med Rep. 2013;8(1):103–12.
Tsung A, et al. HMGB1 release induced by liver ischemia involves Toll-like receptor 4 dependent reactive oxygen species production and calcium-mediated signaling. J Exp Med. 2007;204(12):2913–23.
Su F, Schneider RJ. Hepatitis B virus HBx protein activates transcription factor NF-kappaB by acting on multiple cytoplasmic inhibitors of rel-related proteins. J Virol. 1996;70(7):4558–66.
Lawrence T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol. 2009;1(6):a001651.
Tarn C, et al. Hepatitis B virus X protein activates the p38 mitogen-activated protein kinase pathway in dedifferentiated hepatocytes. J Virol. 2002;76(19):9763–72.
Authors’ contributions
JYJ and SHL designed the study, performed the statistical data analysis, and drafted the manuscript. EKK performed the confocal scanning, histologic analysis and the interpretation of the data. YMH and JKB performed real-time PCR analysis and ELISA. HYK and SKL evaluated clinical data of patients and collected the liver sample. JYJ and CML and JYC participated in the design and coordination of the study. SHL and CML helped to write the manuscript. All authors read and approved the final manuscript.
Acknowledgements
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (2013R1A1A2011680). This study was supported by a grant of the Korean Health Technology R&D Project, Ministry for Health & Welfare, Republic of Korea (HI14C3417).
Compliance with ethical guidelines
Competing interests The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Jhun, J., Lee, S., Kim, H. et al. HMGB1/RAGE induces IL-17 expression to exaggerate inflammation in peripheral blood cells of hepatitis B patients. J Transl Med 13, 310 (2015). https://doi.org/10.1186/s12967-015-0663-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-015-0663-1